ゲノム配列が既知の生物種であり、cDNAやESTを得られたのであれば、そのゲノム配列上のエクソン・イントロン構造を予測することができます。これは転写開始点を同定したり、スプライシングバリアントを解析する前段階と言えます。
【計算時間と精度】このようなエクソン・イントロン構造の予測は、cDNAやESTの配列をゲノム配列に対してアライメントすることにすぎませんが、アライメント中のギャップとしてイントロンの存在が許されるようにアルゴリズムが拡張されており、単純なアライメントと比べて、スプライスサイトのコンセンサス配列やイントロンの長さ等、考慮することが増えるため、比較的に計算時間がかかってしまいます。また、いろいろと考慮してアライメントを生成するといっても、現実を完全に再現するには至っていませんので、正しい予測を行うためには少し工夫が必要です。
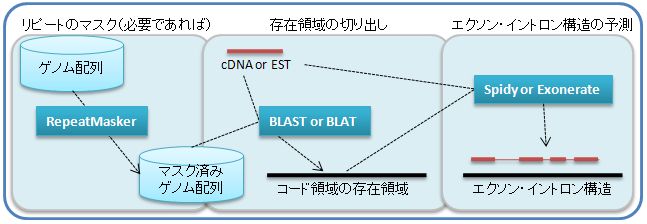
【計算手順の概要】以下に、計算手順の概要を示します。
- リピートを持つ生物種を対象とする場合、RepeatMaskerを用いてゲノム配列中のリピート配列をマスクします。(マスクとは、アライメントされないように、配列中の文字をNやXに置き換えることです。マスクに使用される文字はツールによりますのでツールのマニュアルを確認してください。)リピート配列をマスクすると、アライメントにかかる計算時間が激減します。
- BLASTやBLATといった高速なアライメントツールを用いて、だいたいの存在領域を求めます。そして、ゲノム配列からその領域を切り出します。このように存在領域を限定することは、計算速度の向上だけでなく精度の向上にもつながります。存在領域を制限しておかないと、現実よりも大きなイントロンが入ってしまうことがあります。このような場合、存在領域を再検討して配列を切り出し直すか、イントロン長のパラメータを再設定する必要があります。cDNAやESTもしくはゲノム配列の配列決定精度が若干でも低いと上述のような現象が起こりえます。
- SpideyやExonerateを用いてエクソン・イントロン構造を予測します。
 【WWWサイトの利用】
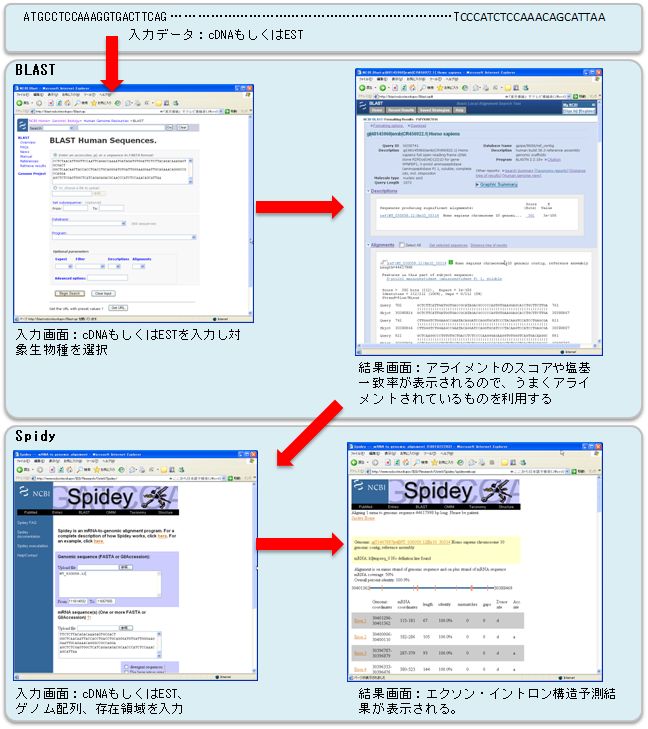
【WWWサイトの利用】BLASTやBLAT、Spideyはインターネットを通じて利用可能です。以下に、BLASTとSpideyのWWWサイトを利用して、エクソン・イントロン構造を予測する手順について説明します。
- BLASTのWEBサイトにアクセスします。幾つかの生物種に関してはゲノム配列データベース("BLAST Assembled Genomes")が整備されていますので、もし興味対象の生物種がそこにあれば、それ用のBLAST実行画面へ行きます。そうでなければ基本的なBLAST("Basic BLAST")の実行画面へ行きます。そして、手持ちのcDNAもしくはESTを入力し、サブミットします。
- BLAST検索の結果、ヒットするものが無ければ、興味対象である生物種のゲノム配列がデータベース中に存在していないかもしれません。その場合には、自分の計算機環境にBLASTやBLATをインストールして、そこで計算を行う必要があります。この場合、ゲノム配列を自分でマスクする必要があるかもしれません。
- SpideyのWEBサイトにアクセスし、手持ちのcDNAもしくはESTと、ゲノム配列とを入力します。このWEBサイトでは、ゲノム配列における遺伝子存在領域を設定することが可能なため、わざわざゲノム配列から切り出す手間を節約することができます。ここには、BLASTによってアライメントされた領域に少し余裕を持たせて(つまり広めに)設定しておくとよいでしょう。この他に、アライメントの最小塩基一致率や、取り扱われる生物種のタイプ(脊椎動物やショウジョウバエ等で、スプライスサイトの予測に影響します)を設定し、サブミットします。
- 得られたエクソン・イントロン構造を見てうまく予測されているようでしたら終了です。もし、cDNAの全長がアライメントされていないとか、アライメントの塩基一致率が低い領域があるとかといったように、予測がうまくいっていないようであれば、設定を変えて再計算するとうまくいく場合があります。
使用できるツール: